Już 21 czerwca poznańska Cytadela zamieni się w morze błękitu. Z okazji Światowego Dnia SLA pojawi się tam 800 flag. Każda z nich to symbol pamięci i wsparcia dla osób zmagających się ze stwardnieniem zanikowym bocznym (SLA), nieuleczalną chorobą, w której dzień po dniu traci się sprawność. Jeśli znasz kogoś, kto choruje, możesz zgłosić go do akcji. Wydarzenie odbywa się w Polsce po raz pierwszy i jest inspirowane międzynarodową inicjatywą, odbywającą się co roku w Waszyngtonie. Redakcja Pacjenci.pl objęła patronat nad tym wydarzeniem. 21 czerwca w poznańskim parku Cytadela powstanie instalacja 800 błękitnych flag dla chorych na SLAKażda flaga to imię konkretnej osoby i hołd dla zmagających się z tą ciężką, paraliżującą chorobąBliskich można zgłaszać do akcji poprzez stronę www.21czerwcasla.pl w terminie do 10 czerwca br
Mówi się, że neurofibromatoza to najczęstsza z chorób rzadkich. Według ostatnich badań, średnio jedno na 2,5 tys. rodzących się dzieci przychodzi na świat z tą genetyczną wadą, która powoduje niekontrolowany rozrost guzów na ciele i narządach wewnętrznych. „Nasze życie przypominało życie na tykającej bombie. Córka mogła pójść spać normalnie, a obudzić się z porażeniem czterokończynowym” – wyznaje Adam Szargut, chorujący na neurofibromatozę i zarazem ojciec chorej Paulinki, która dzięki operacji i nowoczesnej terapii jest dziś w dobrej kondycji. Z okazji Światowego Dnia Neurofibromatozy (17 maja) rozmawiamy o kolejkach do specjalistów, braku leczenia dla dorosłych i potrzebie opieki koordynowanej dla nich. Neurofibromatoza to rzadka choroba genetyczna, która powoduje niekontrolowany rozrost guzówDzieci mają dostęp do terapii i opieki koordynowanej (CEKOM), dorośli są ich pozbawieniTerapia lekiem selumetynib zmniejsza guzy, jednak dorośli w Polsce wciąż nie mają do niej dostępu

Orzeczenie o niepełnosprawności to dla wielu osób jedynie dokument potwierdzający stan zdrowia. Jednak w 2026 roku kod 08-T otwiera drzwi do konkretnego wsparcia finansowego, ulg w pracy oraz ułatwień w życiu codziennym. Co dokładnie kryje się pod tym symbolem i jak wycisnąć z orzeczenia to, co najcenniejsze? Wyjaśniamy.Co oznacza kod 08-T?Znaczny stopień: Najszerszy pakiet wsparciaUmiarkowany stopień: Wsparcie dla pracownikaLekki stopień: Podstawowe ulgiWażne dla pracowników w 2026 roku
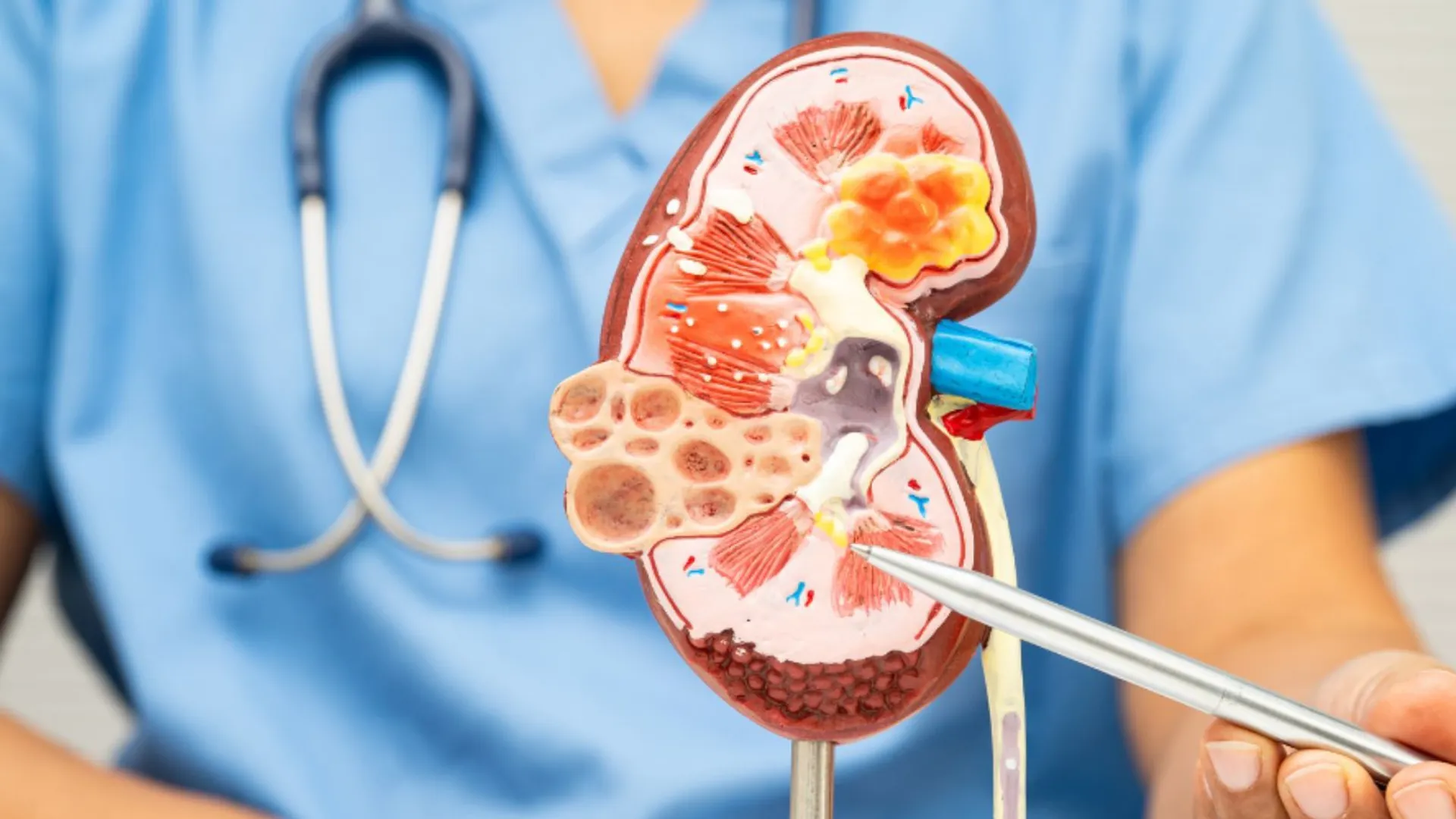
Ból nerek, nawracające kamienie, kolejne wizyty u lekarza i diagnoza kamicy moczowej. Pierwotna hiperoksaluria, ultrarzadka choroba genetyczna, przez lata potrafi ukrywać się pod maską „zwykłej” kamicy, aż w końcu prowadzi do nieodwracalnej niewydolności nerek, uszkodzeń wielu narządów, a w skrajnych przypadkach zagrożenia życia. O tym, dlaczego tak trudno ją rozpoznać opowiada prof. dr hab. n. med. Przemysław Sikora.Pierwotna hiperoksaluria to rzadka choroba genetyczna, która latami podszywa się pod zwykłą kamicę moczową, co opóźnia diagnozę i prowadzi do nieodwracalnego zniszczenia nerek oraz powikłań wielonarządowychChoć źródłem problemu jest defekt metaboliczny w wątrobie (produkującej nadmiar szczawianów), narząd ten pozostaje strukturalnie zdrowy, podczas gdy toksyczne związki niszczą nerki, które nie nadążają z ich filtrowaniemW Polsce dostępny jest już nowoczesny lek oparty na tzw. interferencji RNA, który hamuje postęp choroby, zastępując konieczność wykonywania ryzykownych, jednoczesnych przeszczepów wątroby i nerki

To może być zaskoczenie dla tysięcy pacjentów. Ministerstwo Zdrowia opublikowało komunikat, z którego wynika, że aż 89 leków traci finansowanie ze środków publicznych. Chodzi o terapie stosowane m.in. w leczeniu nowotworów, chorób rzadkich i neurologicznych. Dla części chorych oznacza to pilną konieczność sprawdzenia, czy będą mogli kontynuować leczenie.• Ministerstwo Zdrowia publikuje nową listę leków bez finansowania• Z wykazu wypadają terapie stosowane w ciężkich chorobach• Co z pacjentami, którzy już rozpoczęli leczenie?• Zmiany wymagają szybkiej reakcji ze strony chorych i lekarzy

Stwardnienie zanikowe boczne przez lata było chorobą, w której medycyna mogła jedynie spowalniać nieuchronny proces. Dziś sytuacja zaczyna się zmieniać – pod warunkiem, że pacjent zostanie odpowiednio szybko i kompleksowo zdiagnozowany. Neurolodzy mówią wprost: badania genetyczne powinny stać się standardem u każdego chorego na ALS, bo od nich może zależeć dostęp do leczenia, które realnie wpływa na przebieg choroby.ALS – rzadka choroba o dramatycznym przebieguDlaczego geny mają dziś kluczowe znaczenie?Leczenie celowane zmienia perspektywę chorychNowe polskie wytyczne mają skrócić drogę do diagnozy

Dostęp do rehabilitacji pulmonologicznej, łatwiejszy dostęp do specjalistów na każdym etapie choroby oraz profilaktyka, która jest skuteczna, a nie tylko na papierze – o to apelują pacjenci z przewlekłą obturacyjną chorobą płuc (POChP). Chorzy, ich opiekunowie i lekarze podkreślają, że system wciąż nie nadąża za potrzebami pacjentów, którzy w walce o własny oddech, nie mają sił i możliwości, by walczyć o swoje. POChP dotyka 2 mln Polaków i jest trzecią przyczyną zgonów, lecz z powodu rzadkiego wykonywania spirometrii często diagnozowana jest zbyt późnoSystem opieki jest niewydolny: pacjenci nie mają dostępu do rehabilitacji pulmonologicznej (tylko 20 ośrodków w kraju) i czekają w długich kolejkach do specjalistówSzansą dla chorych są nowoczesne leki biologiczne i trójskładnikowe, które hamują postęp choroby i zmniejszają ryzyko groźnych dla życia zaostrzeń

Opieka koordynowana dla pacjentów z neurofibromatozami (NF) ma szansę w przyszłym roku wejść do katalogu świadczeń gwarantowanych Ministerstwa Zdrowia. Oznaczałoby to zakończenie trwającego od pięciu lat i trzykrotnie przedłużanego pilotażu oraz zapewnienie chorym w każdym wieku systemowego wsparcia. Dla pacjentów, szczególnie dorosłych, to moment przełomowy.Pilotaż koordynowanej opieki nad pacjentami z neurofibromatozą (NF) jest pozytywnie oceniany i ma szansę wejść do świadczeń gwarantowanych, co zapewni pacjentom kompleksową opiekę, choć nadal brakuje w niej systemowego wsparcia dla dorosłych chorychNeurofibromatoza określana jest jako "tykająca bomba" o nieprzewidywalnym przebiegu, od niegroźnych zmian skórnych po monstrualne guzy uciskające narządy, dlatego kluczowa jest stała opieka w wyspecjalizowanych ośrodkach, które mogą wcześnie wykryć i leczyć powikłaniaPrzełomem w leczeniu dzieci z NF1 jest refundowana od 2024 r. terapia selumetynibem, która u 2/3 pacjentów przynosi poprawę, a u reszty zatrzymuje postęp choroby; trwają starania o rozszerzenie dostępu do tego leczenia także dla dorosłych pacjentów

Polski program lekowy B.102 dotyczący leczenia rdzeniowego zaniku mięśni (SMA) stał się modelem, na którym wzorują się inne kraje europejskie. Po siedmiu latach jego funkcjonowania widać wyraźnie, że decyzja o refundacji pierwszej na świecie terapii – nusinersenu, była punktem zwrotnym nie tylko w rokowaniach pacjentów, ale i w budowie nowoczesnego systemu opieki nad chorymi z SMA. Dane z praktyki klinicznej potwierdzają trwałość i poprawiającą się skuteczność terapii, a Polska znajduje się dziś w czołówce państw, które zapewniają kompleksowy dostęp do leczenia tej rzadkiej choroby.Program lekowy B.102 to modelowy system leczenia SMA, na którym wzorują się inne kraje europejskie — obejmuje wszystkich pacjentów, niezależnie od wieku i typu choroby, zapewniając pełny dostęp do terapii i możliwość jej zmiany w razie potrzebyPonad 70% pacjentów leczonych nusinersenem uzyskało klinicznie istotną poprawę, a u wszystkich zatrzymano postęp choroby; efekty terapii rosną wraz z czasem jej stosowania, co potwierdzają dane z siedmioletniej obserwacji w PolscePolska należy do liderów w Europie w diagnostyce i leczeniu SMA — dzięki ogólnokrajowym badaniom przesiewowym noworodków i kompleksowej opiece terapeutycznej, kraj ten wyznacza standardy w leczeniu chorób rzadkich

W Polsce żyje ponad trzy miliony osób zmagających się z chorobami rzadkimi. Najczęściej każda z tych chorób dotyka stosunkowo niewielkiej grupy chorych, jednak razem tworzą oni prawie jedną dziesiątą społeczeństwa. Najnowszy, piąty już raport „Audyt Pacjencki Krajowego Forum ORPHAN 2025” pokazuje, jakie potrzeby pacjentów z chorobami rzadkimi są nadal niezaspokojone oraz jakie są wyzwania stojące przed polskim systemem ochrony zdrowia.Pacjenci domagają się ustawy – 100% organizacji wskazuje na konieczność uchwalenia Ustawy o Chorobach Rzadkich i stworzenia spójnej strategii opiekiLeki i diagnostyka wciąż poza zasięgiem – 97% docenia zmiany w refundacji leków sierocych, ale 70% wciąż apeluje o lepszy dostęp do nowych terapii; 96% sygnalizuje problemy z diagnostykąWsparcie wykraczające poza medycynę – 96% organizacji podkreśla potrzebę opieki psychologicznej, 95% edukacji, a 91% lepszego wsparcia socjalnego; większość krytykuje obecny system orzekania o niepełnosprawności

W Polsce żyje około 400 dzieci cierpiących na dystrofię mięśniową Duchenne’a. To rzadka, genetyczna choroba, która prowadzi do postępującego i nieodwracalnego zaniku mięśni. Dotyka wyłącznie chłopców – z czasem odbiera im możliwość poruszania się, a ostatecznie także samodzielnego oddychania.Nadzieję daje nowa terapia genowa opracowana w USA – pierwszy krok ku spowolnieniu choroby. Jej koszt jednak jest ogromny.
Na dźwięk słowa „hemofilia” większość z nas wyobraża sobie małego chłopca, który krwawi z nosa. Takie krwawienia faktycznie mogą występować w przebiegu tej rzadkiej choroby, lecz nie są one standardem. Wielu chorych mierzy się z jeszcze bardziej dotkliwymi, choć niewidocznymi gołym okiem objawami – krwawieniami do stawów lub do mięśni.
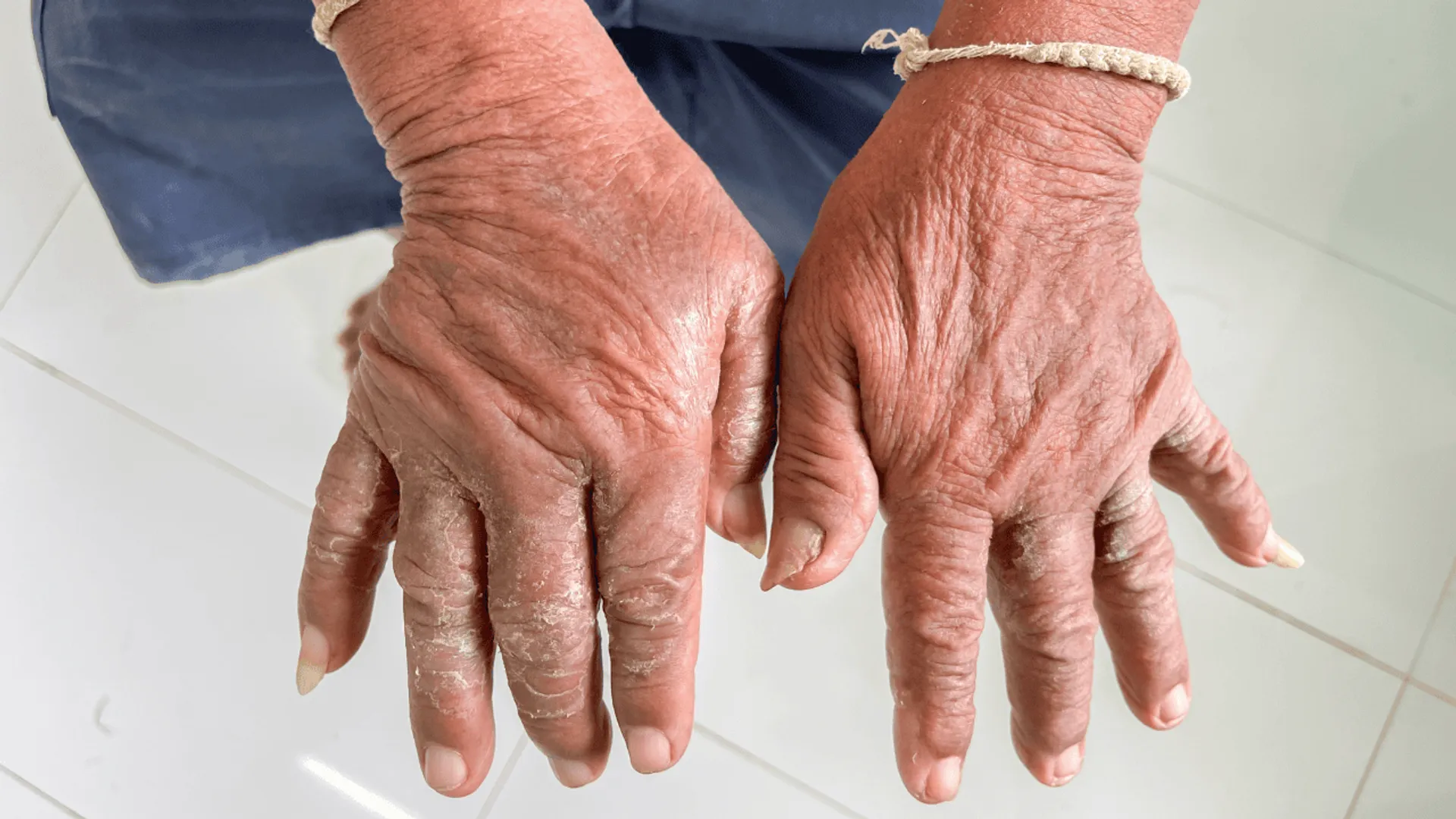
Twardzina układowa (łac. scleroderma) to rzadkie, przewlekłe schorzenie tkanki łącznej o podłożu autoimmunologicznym, charakteryzujące się nadmiernym odkładaniem kolagenu w skórze i innych tkankach. Choroba wpływa na wiele narządów i układów w organizmie, w tym na skórę, naczynia krwionośne, przewód pokarmowy, płuca, serce i nerki. W Polsce choruje na nią ok. 10 000 osób, natomiast w ciągu roku występuje 4–12 nowych zachorowań na milion mieszkańców.Tkanka łączna występuje w całym organizmie, a jej funkcją jest spajanie tkanek i zapewnianie podpory narządom. W przebiegu twardziny układowej dochodzi do postępującego włóknienia skóry (w przypadku twardziny ograniczonej) i narządów wewnętrznych (w twardzinie uogólnionej), co skutkuje zaburzeniami w ich funkcjonowaniu.

29 lutego, szefowa resortu zdrowia Izabela Leszczyna zadeklarowała, że 1 kwietnia ukaże się nowa lista leków refundowanych, która obejmie większą część chorych. Jest to dobra wiadomość dla szczególnej grupy pacjentów.